5 Montagem de sequências
A primeira etapa computacional de uma análise filogenética é a montagem das sequências (sequence assembly) de cada um dos terminais a partir dos cromatogramas de cada primer sequenciado.
5.1 Cromatogramas
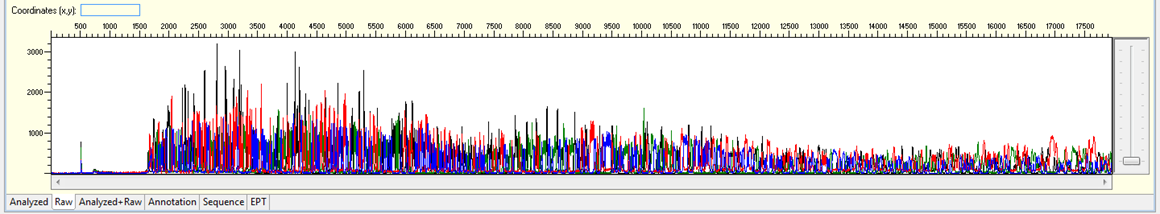
5.1.1 Conceito
- Rastro (trace) do sinal dos nucleotídeos marcados por corante de acordo com a intensidade, indicada por quatro cores correspondentes às quatro bases nitrogenadas.
5.1.2 Finalidade
- Verificar a qualidade do sinal
- Detecção dos picos
A baixa qualidade do sinal é denominado de ruído (noise). O ruído é caracterizado por picos pouco acentuados, com altura mais baixa e mais largos.
5.1.3 Qualidade do sinal
- Alta
- Sem ambiguidade
- Picos adequadamente distantes com espaços regulares
- Sem ruído
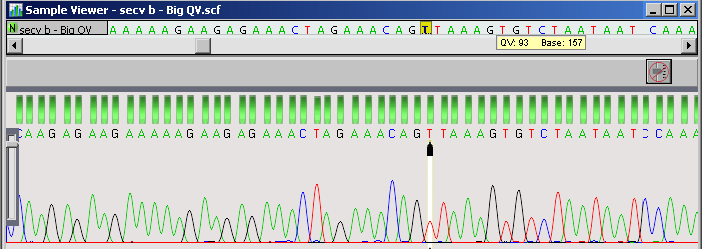
- Média
- Algumas ambiguidades
- Picos adequadamente distantes com espaços regulares
- Alguns trechos homopoliméricos não estão bem resolvidos
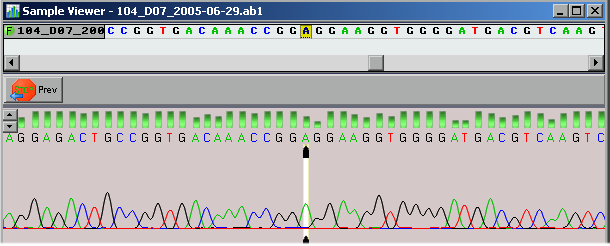
- Baixa
- Algumas ambiguidades
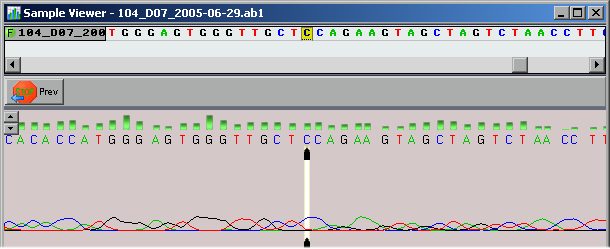
5.1.4 Arquivo .ab1
O arquivo .ab1 contém o eletroferograma da sequência de DNA, bem como dados brutos e outras informações.
O arquivo .ab1 são gerados pelos sequenciadores da Applied Biosystems. Outros sequenciadores utilizam arquivos nos formatos .scf (Standard Chromatogram Format) e .ztr.
Dois programas foram desenvolvidos especialmente para visualizar os cromatogramas:
… mas diversos programas podem realizar a tarefa.
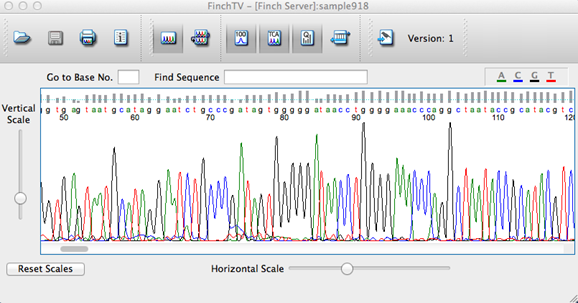
5.1.5 Software FitchTV

O FinchTV 1.4 é um programa open-source para visualização da qualidade e intensidade dos sinais obtidos. Com ele, é possível analisar os dados brutos do cromatograma (raw data).
5.1.6 Manual do sequenciamento Sanger
Diversos fatores podem resultar em baixa resolução do sinal. Observe o capítulo 8 (Troubleshooting) do guia da Applied Biosystems para entender alguns dos principais problemas no sequenciamento:
5.1.7 Interpretação do cromatograma
O processo automatizado de detecção de contigs através do algoritmo base calling de um software é sujeito à erros. E não é raro ocorrerem. Isso deve-se à baixa qualidade do sinal em certas regiões do cromatograma. A qualidade do sinal varia em função de inúmeros fatores e, geralmente, está relacionada à etapa das reações de sequenciamento. Compostos secundários, concentração inadequada de DNA amplificado, presença de contaminantes ou impurezas, ou qualquer outra razão misteriosa, pode atrapalhar as reações de sequenciamento, levando a uma baixa qualidade do sinal. Portanto, o filogeneticista precisa revisar visualmente e manualmente toda a sequência montada automaticamente por um software. Ademais, também é da própria natureza da tecnologia de sequenciamento Sanger, que a qualidade e resolução do sinal começa baixa, mas vai gradativamente melhorando a partir do início da reação, porém começa a decair após um determinado ponto. Consequentemente, a depender da qualidade do cromatograma, a duração dessa tarefa de montagem das sequências pode variar.
Ademais, após o alinhamento múltiplo das sequências, é preciso reconferir os cromatogramas das sequências montadas. Por isso, é fundamental salvá-las no software utilizado. Cada software salvará como um arquivo específico, que não pode ser lido por outros programas. A tarefa de reconferir as montagens após o alinhamento múltiplo é trabalhosa, mas essencial para a correta codificação das homologias moleculares.
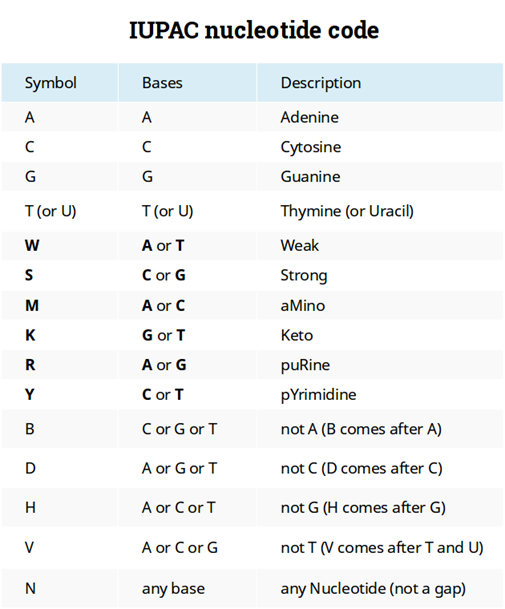
Como decorrência do problema eventual de baixa resolução no cromatograma em algumas posições, um código de ambiguidade de nucleotídeos foi estabelecido pela IUPAC durante o Comitê de Nomenclatura da União Internacional de Bioquímica (NC-IUB), em 1984 (Cornish-Bowden, 1985).
5.1.8 Prática
Para essa atividade será preciso baixar o FitchTV.
Vamos abrir alguns arquivos do cromatograma da região trnD-T de um organismo desconhecido (species_1):
Observe o raw data dos cromatogramas:
- Clique na aba
View->Raw Data; - Na janela que se abre, arraste o
Horizontal Scaletotalmente pra esquerda.
- Clique na aba
Há arquivos .ab1 nesse site: https://www.vanderbilt.edu/wolbachiaproject/lab-5-dna-sequences.
5.2 Alinhamento de contigs
Contigs
Conjunto de sobreposições entre as fitas-simples oriundas de cada primer para formar uma sequência de consenso.
5.2.1 Assembly softwares
A partir do rastro da intensidade dos sinais de cada uma das quatro bases nucleotídicas (raw trace), um programa de computador processa através do processo denominado base calling, que reconhece os picos nas intensidades do sinal de cada base.
Dentre os programas que realizam o base calling para montagem das sequências estão: Geneious, Sequencher, CodonCode Aligner, ChromasPRO, DNA Baser; mas também os open-source: Staden Package e o pacote Phred/Phrap/Consed.
Existem diversos algoritmos para montagem de sequência , dentre os quais, destaca-se: CAP3, PHRAP, TIGR. Os algoritmos de montagem de genoma são conjuntos de procedimentos bem definidos necessários para reconstruir sequências de DNA a partir de dois ou um grande número de fragmentos mais curtos de uma sequência de DNA. Os fragmentos são alinhados uns ao lado dos outros pelo reconhecimento das regiões que apresentam sobreposição (contigs). Essas regiões de contigs são identificadas e mescladas. Algoritmos para reconstruir o genoma completo a partir de milhares de fragmentos como na tecnologia next-generation também têm sido desenvolvidos.

5.2.2 Prática
Para essa atividade será preciso baixar um dos programas que realizam essa tarefa. Contudo, esses programas possuem licenças pagas. Entretanto, as empresas oferecem um período de teste com todas as funcionalidades ativas. Sugestão para essa atividade: Sequencher, Geneious ou CodonCode Aligner.
Vamos utilizar os arquivos listados abaixo, que compõem quatro primers sequenciados do locus trnD-trnT de um organismo desconhecido (species 1):
Siga o passo-a-passo:
- Abra esses arquivos no programa escolhido.
- Execute a função para realizar a montagem dos primers (assemble).
- Inspecione visualmente a montagem dos primers do início ao fim.
- Interprete os cromatogramas e as respectivas sequências e edite manualmente os erros encontrados.
- Finalmente, teremos a sequência consenso montada.
- Não deixe de salvar o arquivo da montagem no
softwareutilizado. Após a etapa do alinhamento, o filogeneticista terá de reconferir essas montagens.
Compare a sua montagem com esta, realizada no programa
Sequencher:
Faça também a montagem do ITS:
Compare a sua montagem do ITS com esta, realizada no programa
Sequencher:
Monte as sequências usando os arquivos .ab1 em: https://www.vanderbilt.edu/wolbachiaproject/lab-5-dna-sequences.